 accredited inspection body for API GMP audits")

审计 本词源于拉丁语“audire”,意谓“去听、去审理”
美国联邦医疗器械法规或ISO标准中对审计有这样的定义:
美国FDA医疗器械管理规范21CFR820总则
“质量审计是指按照规定的时间间隔对制造商的质量系统进行的系统的、独立的检查,以确定质量体系运转及其结果符合质量体系的程序要求,并确保质量体系程序得到有效实施并适合于实现质量体系的目标。”
ISO 9000, ISO 19011
“为获得审计证据并对其进行客观评价,以确定其对审计准则的依从性进行的系统的、独立的并形成文件的过程。”
检查 本词同样源于拉丁语Inspicere意谓 “查看、检查”。
欧盟2001/20/EC指令对临床试验对检查有如下定义:
“主管机构对文件、设施记录、质量保证措施以及主管机关认为与临床试验有关的,且位于试验场所,或在赞助商和/或合同研究机构的设施内,或位于主管当局认为适合接受检验的其他任何设施内的其他任何资源所做的正式审核。”
由此可见,审计与检查两个词语都描述通过系统性程序来检查并评估企业的质量体系是否符合法规要求以及其符合程度。“审计”这个词属于一般通用术语,可用于各种类型的评估;而“检查”一词则更为严格,在制药行业中,特是指由主管卫生当局,如欧盟成员国主管卫生当局或欧洲药物质量管理局执行的现场检查。
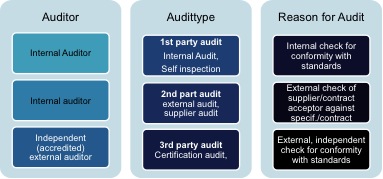
审计通常按照审计原因再进一步分类为“第一方审计”、“第二方审计”和“第三方审计”;如下图显示第三方审计代表了最高的对立性。
按照国际GMP指南规定,如欧盟GMP指南第二部分(=ICH Q7)第二章或第一部分第九章中;明确要求原料药和成品药的生产商应定期进行内部审计或自检。在2013年第十一期《中国医药经贸》“实务”专栏“欧盟GMP深度解读”里也提到过内部审计的重要性。通过调查在自检中发现的缺陷,以及后续跟进的预防与纠正措施 (CAPAs)是否有效落实,能够帮助企业提高GMP依从性。
从笔者的经验来看,由制药企业内部人员定期执行内部审计一般不成问题,然而企业GMP执行力度之高低对审计执行过程以及审计结果的价值有一定影响,这与审计员的资质、其独立性、审计流程、缺陷判断以及后续缺陷的跟进工作有关。说明见下图:
审计员资质 | 审计员独立性 | 审计流程 | 缺陷的评价 | 缺陷的跟踪 | |
GMP执行力弱(低水平) | 弱 | 企业/部门 内部审计员 | 无组织, 安排混乱 | 没评价、总结缺陷措辞不清 | 弱 |
GMP执行力强(高水平) | 强 (高素质而经验丰富) | 外部审计员 | 有组织、 安排有条理 | 有评价/能清晰描述缺陷并做出正确分类 | 强(系统地调查缺陷根本原因以及严格执行合适的整改与预防措施) |
什么是“良好审计规范”?审计注意事项有哪些?
在审计中,审计员与被审查方的行为是密切关联的,这些行为在“良好审计规范”中做出了一般性说明,但当中并没有明确划分审计员与被审查方的角色。在接下来的段落里,我会就现场检查时被审查方应该注意的事项进行进一步的说明。
在审计过程中,审计员应引导审计过程并作开放性提问,如“谁?什么时候?怎样?或:请解释….等等”,之后要等待听取回答或反馈,这个步骤是非常重要的。审计员应小心地记录被提问人员的回复,并根据其它审计时收集的证据(如缺陷和文件内容等)一起做出评估。为避免有任何误解,审计证据的评估也可以视为对理解的再次确认,如“我这样理解对吗? 你的解释是什么意思?”;特别在审计时所用的语言不是审计员或被审查方的母语时,或者这语言同时都不是双方的母语的情况下,再次确认则更为重要,笔者建议审计员需要再次确认。
- 第一步: 证据 (文件、缺陷)
- 第二步: 评估、确认;再次确认,排除误解
- 第三步: 做文字记录
- 第四步: 评级、分类
- 审计报告: 参照审核准则,使用正确的措辞来描述审计发现
评审结束后得出来的审计结论可能符合审计准则、部分符合或是不符合。如果是不符合时,这些不符合项都会按照其严重性来评级和分类,如一般/其他缺陷、主要缺陷、严重缺陷,这种分类对于审计员与被审查方的后续跟进工作是很有用的。典型的“严重缺陷”(等同不符合项)是罕见的,是指严重违反GMP规范而对病人的健康造成潜在的极大危害。严重缺陷通常是在无菌原料药或药物产品的生产上发现、或证实伪造数据、或存在其他欺诈行为的情况下认定的。“主要”和“一般/其他” 缺陷可以根据违反GMP准则和/或对患者构成健康影响的严重程度来区分。通常情况下,同类型的缺陷反复地或系统性地在审计过程中被发现,这样可以归类为一个“主要缺陷”。对于审计发现的分类的进一步说明,可以参考本文末尾欧洲药品管理局(EMA)指南文件中“检查与符合性”的有关定义。
现场检查注意事项
如前文中提到的,欧盟各国主管当局对生产制造商的定期检查被认为是维护和支持GMP符合性的重要手段。检查的结果可能是“符合GMP标准”或是“不符合GMP标准”;对后者而言,其结果可能对被检查企业造成严重影响,例如药物进口欧盟和/或在欧洲销售被限制了。面对官方卫生主管部门(如欧盟/美国FDA/ EDQM检查)的检查时,应该如何准备?在检查过程中,哪些事情应该做,哪些事情应该避免去做?又有哪些事情可以建议大家作为一般规则去遵守呢?
现场检查应对
GMP依从性是一个常规行为,无论什么时候都应该严格遵守GMP规范。因此,一家对GMP执行力很强的企业,并不需要专门为即将到来的官方检查做出额外的准备工作。然而,一些前期的准备,如与检察组就日程安排(日期和时间)、交通与住宿安排、检查范围与需要检查的区域,是否需要配备翻译、哪些文件需要检查等方面提前进行沟通是需要和是有意义的。所有参与检查的企业人员应该是公开的、诚实的并清楚地作出表达,这样可以保持检察官对厂家的信任和信心。如有任何误解或不确定性,应尽快加以澄清。对潜在的缺陷立即采取行动,往往被视为积极的态度并受到检察官的赞赏。为了避免检察官有任何不良印象,包括不合作行为或刻意隐藏某些事情等,被检查的文件/记录、区域以及人员应按要求提前并合理地安排好。
最后一点,当检查员在提问时,只需负责相关工作的人员作出回答,因为他们最清楚自己的工作流程,这也可以很好的证明员工对GMP有深刻的理解。反之,不是负责该工作的人不应该尝试回答问题,因为这样是不恰当的;但可以试着做出说明,如“对不起,这个不是我的职责,所以不是由我来回答您的问题,但某某可以”。 另外,要有针对性地回答提问,回答时需有重点,没有提问的就不应该回答。
现场检查禁忌
在任何情况下,被检查的企业是不应该在现场检查之前做最后的变更或者是重新改造,除非这些是必须的,而且是提前计划好的。以隐瞒事实真相为目的,而不是为了改善和解决问题的任何行为,笔者都明确建须要避免,因为这些行为都会引起检察官的怀疑,从而导致整个现场检查的氛围变得紧张起来。针对这点,常常看到的行为包括中断生产、墙壁喷漆、重新替换一次性材料(如垫片、橡胶软管)等等、变更/更新标签、或重写文件/记录;而这一切在现场检查前或检查进行中都不被视为有利行为。
作者:
Stefan Kettelhoit博士,Blue Inspection Body GmbH(以下简称"BI")总经理。BI是一家取得官方认可资质的独立第三方检查机构,在全球范围内开展原料药、 药用辅料、药品以及其他各类良好规范(G x P)的审计。2013年6月1日B I 与中国医药保健品进出口商会建立了合作关系,共同协助国内药品与原料 药生产商提升质量管理系统,达到欧盟GMP要求。公司网址:www.blue-inspection.com
参考文件:
欧洲药品管理局“有关检查和信息交流的共同体程序编制总文件,检查与符合性”2013年6月27日EMA/385898/2013 Rev 16