 accredited inspection body for API GMP audits")

Audit – the word is derived from the latin language word “audire”. It means to hear, to listen.
Definitions can be found in the US Code of Federal Regulations for Medical devices or the ISO standard as follows: „Quality audit means a systematic, independent examination of a manufacturer’s quality system that is performed at defined intervals and at sufficient frequency to determine whether both quality system activities and the results of such activities comply with quality system procedures, that these procedures are implemented effectively, and that these procedures are suitable to achieve quality system objectives.”(21CFR820 – Medical devices, General provisions)
„Systematic, independent and documented process for obtaining audit evidence and evaluating it objectively to determine the extent to which audit criteria are fulfilled.” (ISO 9000, ISO 19011 )
Inspection – the word is also derived from latin language and means to look at, to inspect.
A definition (related to clinical trials) can be found in the EU Directive 2001/20 as follows: „The act by a competent authority of conducting an official review of documents, facilities, records, quality assurance arrangements, and any other resources that are deemed by the competent authority to be related to the clinical trial and that may be located at the site of the trial, at the sponsors and/or contract research organisations facilities, or at other establishments which the competent authority sees fit to inspect.” (DIRECTIVE 2001/20/EC)
It thereby becomes clear, that both terms describe a systematic process of evaluating and examinating systems being in place for organizations and judging the degree of conformance to accepted or legally required standards. In this sense the term “audit” is the more general term, which is used for every kind of such evaluation, whereas the term “inspection” is in a more strict (pharmaceutical) way only used conducted for on-site audits conducted by official health authority bodies ( e.g. EU or other national competent health authorities (NCAs) or the European Directorate for the Quality of Medicines & HealthCare, EDQM).
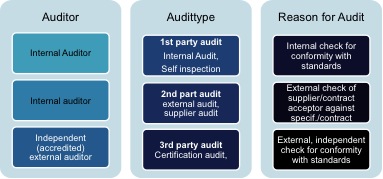
For audits, frequently the type of audit altogether with the reason for such audit is further distinguished. This is done with the terms “1st party”, “2nd party” and “3rd party” audit, whereas the latter represent the highest degree of independence as can be derived from the following picture:
The conduct of internal audits (also referred to as “self inspections”) is legally required for the manufacturers of APIs and finished medicinal products according to international GMP guidelines (see for instance EU GMP guideline part II (= ICH Q7) chapter 2 or EU GMP part I guideline chapter 9). As already mentioned in the second publication of this series, this is seen as important means to support and increase the GMP compliance level of an organization by investigating potential deficiencies identified in the audits and subsequent follow-up on suitable corrective and preventive actions (CAPAs).
The conduct of such audits is typically not an issue according to the experiences of the author and regularly done by the persons being in charge for this in pharmaceutical companies. However, the process of how the audits are conducted and thereby the value derived from the audits is broadly differing between “low-level GMP” and “high-level GMP” companies concerning e.g. auditor qualification, auditor independence, audit process, judgement of deficiencies and follow-up on audit findings (see also the following picture).
What is anticipated as “Good auditing practice”? What are Do’s and Don’t’s for audits?
The behavior during audits for the auditor and auditee is closely inter-related. It is hereby only generally described as what is commonly believed as “Good auditing practice” and not specifically distinguished for the role of the auditor and auditee. After that, the next chapter will in more detail cover the Do’s and Don’t’s during inspections related only to the role and responsibility of the auditee (i.e. companies being audited).
During an audit the auditor should manage and steer the audit process by raising open questions (“Who?” “When?” “How?” or: “Please explain . . .” etc.). Afterwards it is important to wait and listen for the answer / feedback from the persons being audited. The answers provided by audited persons should carefully be documented and evaluated altogether with gathered own evidence such as observations or document contents. The evaluation of the audit evidence should also consider a confirmation of the understanding (“Did I get you right with . . . ?”, “What do you mean with your explanation . . . .?” etc.) in order to avoid any misunderstanding or misinterpretation. This is even more important and recommended in cases, where the audit language is not the mother tongue of the auditor and / or auditee or both of them. Only after that, the degree of conformance for the audit evidence with pre-defined audit criteria (e.g. EU GMP part II / ICH Q7 or other) should be assessed as objectively as possible by the auditor (see bullet points hereunder).
- First step: evidence (document, observation)
- Second step: evaluation, confirmation (CAVE: misunderstanding, misinterpretation)
- Third step: documentation
- Fourth step: grading, classification
- Audit report: correct phrasing and reference to audit criteria
The outcome of this evaluation as per each audited item may be conformance, partial conformance or non-conformance. In case of non-conformances a grading / classification into the degree of non-conformance (e.g. “minor / other”, “major”, “critical”) has proven to be useful in order to facilitate the follow-up for the auditor and the auditee. Typically “critical” deficiencies (=non conformances) are rare and subject to severe and systematic GMP violations with potential severe harm to patients’ health. This is often the case for deficiencies found in the manufacturing of sterile APIs or drug products or in case of evidenced data falsification or other fraud actions. “Major” and “minor /other” deficiencies can be distinguished based upon the severity of the GMP violation and / or effect on patients’ health. Often, a “major” deficiency is also justified in cases where the same type of shortfall is recurrently or systematically detected in an audit. Further reference for the classification of audit findings can be derived from the respective definitions provided in the European Medicines Agency (EMA) guideline document “Compliance and Inspection” as referenced at the end of this article.
Do’s and Don’t’s for inspections
As already mentioned earlier, regular inspections of manufacturers by inspectors from NCAs are regarded as important means in Europe to maintain and support the GMP compliance level. The outcome of such inspections for affected companies may be “GMP-compliant” or “GMP-non-compliant” with resulting severe commercial impact in the latter case (e.g. import and / or sales restrictions). What can be recommended as general rules to companies facing official health authority inspections (e.g. EU / US-FDA / EDQM inspections)? How to prepare for such inspections? What to do (and what to avoid) during inspections?
Do’s
GMP readiness and compliance is expected to be there at all time. Therefore, companies with a constant and high level of GMP-compliance implementation do not need to specifically prepare for upcoming inspections with extra work or efforts. However, preparation of an inspection is meaningful by communication upfront with the inspection team and clarifying any questions concerning e.g. travel & accommodation, inspection date & timing (agenda), inspection scope, translation, areas to be inspected, expected documentation before and during the inspection etc. During the inspection all participating persons from a company should behave open, honest and should clearly communicate in order to maintain an inspection situation of trust and confidence. Any misunderstanding or uncertainty should be clarified as soon as possible. Immediate action on potential deficiencies is often regarded as positive attitude and appreciated by inspectors. The availability of persons, documents / records or areas to be inspected should be taken care of as requested and reasonable, in order to avoid any impression for un-cooperative behavior or unwanted hiding of items / things.
Finally, only those persons being responsible (and thereby knowledgable) should answer only to those questions of the inspectors, which have been asked. It is a good attitude and shows high level of GMP understanding, if a person, who is npt responsible for a certain task does not try to answer (often improperly) but instead points out something like :”Please apologize, this is not my responsibility, therefore it is not me to answer your question, but Mr / Mrs X”. Furthermore an answer should focus on the question rather than to spread out to items, which were not asked.
Don’t’s
In the light of an inspection a company is not supposed to do last minute changes and / or reconstructions, unless anyhow required / planned. Any activities suitable to hide items rather than improving and / or solving them are clearly recommended to be avoided, since this may trigger suspicion to the inspectors and move into an unwanted and unsupportive inspection atmosphere. With regard to this, frequently seen things such as interruption of production, painting walls, replacing all kinds of disposable materials (e.g. gaskets, hosepipes etc.), changing / re-newing labels or re-writing documents / records are definitely points, which are not regarded as supportive before and / or during inspections.
References
EMA Compilation of Community Procedures on Inspections and Exchange of Information, Compliance and Inspection, 27 June 2013, EMA/385898/2013 Rev 16: www.ema.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2009/10/WC500004706.pdf